

В последнее время мужское бесплодие стало не менее острой проблемой, чем женское. Наблюдается общая динамика снижения качества спермы у молодых людей. Факторы, которые могут привести к ухудшению показателей спермограммы, разные, но не всем мужчинам с плохой спермограммой показано обследование и исключение генетического фактора, так кому же рекомендуется генетический тест?
Содержание
- 1 ПРЯМЫЕ ПОКАЗАНИЯ К НАЗНАЧЕНИЮ ВРАЧОМ АНДРОЛОГОМ ГЕНЕТИЧЕСКОГО ОБСЛЕДОВАНИЯ – ЭТО?
- 2 КАКИЕ АНАЛИЗЫ НА ГЕНЕТИКУ МОЖЕТ НАЗНАЧИТЬ АНДРОЛОГ ПРИ БЕСПЛОДИИ?
- 3 ДЛЯ ЧЕГО НЕОБХОДИМЫ ДАННЫЕ АНАЛИЗЫ ВРАЧУ АНДРОЛОГУ?
- 4 МИКРОДЕЛЕЦИИ AZF – РЕГИОНОВ (А, B,C) Y-ХРОМОСОМЫ , ЧТО ЭТО?
- 5 ИССЛЕДОВАНИЕ КАРИОТИПА ЧЕЛОВЕКА, ЧТО ЭТО ЗА АНАЛИЗ?
- 6 МУКОВИСЦИДОЗ – МУТАЦИИ ГЕНА CFTR (14 МУ –ТАЦИЙ), ПОЧЕМУ РЕКОМЕНДУЮТ ДАННЫЙ АНАЛИЗ ПРИ МУЖСКОМ БЕСПЛОДИИ?
- 7 Генетический фактор мужского бесплодия
- 8 Роль мужского фактора в зачатии и беременности
- 9 Врожденная патология генетического аппарата
- 10 Выводы
- 11 Генетически обусловленные формы бесплодия у мужчин: основные характеристики и практические аспекты лабораторной диагностики
ПРЯМЫЕ ПОКАЗАНИЯ К НАЗНАЧЕНИЮ ВРАЧОМ АНДРОЛОГОМ ГЕНЕТИЧЕСКОГО ОБСЛЕДОВАНИЯ – ЭТО?
- Наличие в спермограмме выраженной олигозооспермии и / или тератозооспермии (низкое количество сперматозоидов и очень плохая морфология), олигоспермии (с выраженным уменьшением объема эякулята);
- Азооспермия (отсутствие сперматозоидов в сперме);
- Изменение уровня половых гормонов (анализ крови);
- Небольшой объем яичек (уменьшение объема яичек в 2-3 раза меньше нормы);
- Если у супружеской пары в анамнезе были выкидыши (выкидыши или обморожения, особенно на ранних стадиях).
КАКИЕ АНАЛИЗЫ НА ГЕНЕТИКУ МОЖЕТ НАЗНАЧИТЬ АНДРОЛОГ ПРИ БЕСПЛОДИИ?
Следует отметить, что при назначении генетического обследования андролог всегда опирается на данные анамнеза пациента, осмотра и клинического обследования, на основании этих данных могут быть рекомендованы следующие генетические тесты:
- Микроделеции AZF — участки (a, b, c) Y-хромосомы ;
- Кариотип ;
- Ген фактора CFTR муковисцидоза.
ДЛЯ ЧЕГО НЕОБХОДИМЫ ДАННЫЕ АНАЛИЗЫ ВРАЧУ АНДРОЛОГУ?
- Выявление причины мужского бесплодия;
- Прогноз эффективности консервативной терапии (пищевые добавки, поливитамины, аминокислоты, гормональная стимуляция и др.);
- Выбор метода и прогноз эффективности выбранного метода лечения бесплодия (естественный способ зачатия или ЭКО / ИКСИ).
! При выявлении генетического фактора бесплодия может в корне измениться тактика ведения супружеской пары, в некоторых случаях необходимо проведение предимплантационной диагностики плода с целью исключения передачи генетического заболевания потомству.
МИКРОДЕЛЕЦИИ AZF – РЕГИОНОВ (А, B,C) Y-ХРОМОСОМЫ , ЧТО ЭТО?
Данное исследование позволяет установить генетические причины репродуктивных нарушений у мужчин, оценить эффективность лечебных процедур. AZF — участки мужской Y-хромосомы включают большое количество генов, ответственных за производство спермы. Удаление, то есть потеря этих генов, приводит к изменению сперматогенеза. Наиболее изучены AZFа, AZFb и AZFс. Эти мутации обнаруживаются у 11% мужчин с азооспермией и у 8% мужчин с тяжелой олигоспермией.
Период анализа обычно составляет не менее 19 рабочих дней.
Материал исследования: цельная кровь.
ИССЛЕДОВАНИЕ КАРИОТИПА ЧЕЛОВЕКА, ЧТО ЭТО ЗА АНАЛИЗ?
Изучение кариотипа проводится с целью диагностики хромосомных синдромов. В норме у женщин кариотип 46, XX; а для мужчин 46, ху.
Кариотип — это описание хромосом соматических клеток организма (количество, размер, форма, структурные особенности) на метафазной стадии деления клеток. В основе хромосом лежит ДНК, носитель генетической информации. Такой анализ имеет большое значение, позволяя диагностировать ряд хромосомных заболеваний, вызванных как тяжелыми нарушениями кариотипов (нарушение числа хромосом), так и нарушением хромосомной структуры или множеством клеточных кариотипов в организме (мозаицизм). Как правило, нарушения кариотипа у человека сопровождаются множественными пороками развития, большинство таких аномалий несовместимы с жизнью и приводят к самопроизвольным абортам на ранних сроках беременности. Однако довольно большое количество плодов с аномальным кариотипом, около 2,5%, созревают до окончания беременности, и эта патология иногда может быть обнаружена только впервые, например, в репродуктивном возрасте во время беременности. Обследование на предмет бесплодия.
Материал исследования: цельная кровь.
Срок изготовления анализа: не менее 19 рабочих дней.
Например, некоторые заболевания человека, вызванные аномалиями кариотипа: синдром Клайнфельтера; Синдром Шерщевского-Тернера; полисомия по Х-хромосоме; Болезнь Дауна; Синдром Эдварда; Синдром Патау; синдром кошачьего плача
МУКОВИСЦИДОЗ – МУТАЦИИ ГЕНА CFTR (14 МУ –ТАЦИЙ), ПОЧЕМУ РЕКОМЕНДУЮТ ДАННЫЙ АНАЛИЗ ПРИ МУЖСКОМ БЕСПЛОДИИ?
Изучение гена CFTR может иметь диагностическое значение как у пациентов с клиническими проявлениями заболевания, так и прогностическое значение для выявления носителя неблагоприятных мутаций у здоровых людей, вступающих в брак и / или планирующих рожать.
Мутации в гене CFTR являются наиболее частой причиной мужского бесплодия, связанного с односторонней или двусторонней врожденной обструкцией или отсутствием семявыносящего протока. Отсутствие семявыносящего протока наблюдается у 2% бесплодных мужчин и у 6% мужчин с обструктивной азооспермией. Отличительной особенностью спермограмм с различными мутациями гена муковисцидоза являются олигоастенотератозооспермия, изолированная олигозооспермия, азооспермия неизвестного происхождения, уменьшение объема сперматозоидов, отсутствие или низкая концентрация фруктозы, аномальная вязкость эякулята. Из-за врожденной облитерации семенных протоков почти все мужчины с муковисцидозом бесплодны. У женщин, страдающих муковисцидозом, происходит густая цервикальная секреция спермицидов, что снижает вероятность оплодотворения.
Материал исследования: цельная кровь.
Сроки анализа: не менее 19 рабочих дней.
Генетический фактор мужского бесплодия

Неспособность или неспособность зачать ребенка, синдром выкидыша, бесплодие — все это звенья одной цепи, и супружеские пары, у которых нет детей, должны быть обследованы на предмет зачатия. Одним из факторов мужского бесплодия является снижение активности репродуктивной функции, вызванное ранее перенесенными заболеваниями мочеполовой системы, нарушением сперматогенеза или другими генетическими мутациями.
Роль мужского фактора в зачатии и беременности
Полная половая жизнь без использования противозачаточных средств по логике должна приводить к зачатию, но почти у 12% супружеских пар этого не происходит (данные ВОЗ). Здоровье женщины в этом случае играет немалую роль, но если по женской линии проблем нет, то мужчина, помимо обычных анализов, обязан пройти генетическое обследование.
Причины бесплодного брака в 40% кроются в состоянии здоровья мужчины, он также отвечает за беременность супруга, поскольку генетики предполагают, что аборты в первом триместре могут быть спровоцированы «отбраковкой» дефектов эмбриона, т.е изменения которого вызваны мутацией хромосомы Y. Повторяющиеся выкидыши на ранних стадиях — хороший повод заподозрить генетическое бесплодие у мужчин, вызванное различными факторами.
Значительные дефекты сперматозоидов могут не быть видны на спермограмме, поэтому, помимо обычного биохимического теста, рекомендуется провести молекулярно-цитологическое и генетическое исследование для оценки состояния генетического аппарата мужчины.
необходимо знать, что в проблеме мужского бесплодия развитие половины тяжелых форм заболевания обусловлено генетическими причинами, вызванными нарушением сперматогенеза, который обычно является четко контролируемым процессом. Около 2000 генов ответственны за созревание сперматозоидов, и отказ на любом участке приводит к дефектам различной этиологии и сложности, которые, в свою очередь, могут вызывать от небольших изменений активности сперматозоидов до полной дисфункции половых клеток.
Врожденная патология генетического аппарата
Среди генетических факторов мужского бесплодия можно выделить три основных:
— нарушения на уровне хромосомных аберраций;
— изменения на генном уровне (генные мутации);
— поражения на уровне цепи ДНК.
Визуально эти изменения невозможно проследить, поэтому необходима генетическая диагностика с использованием цитогенетических методов, показывающих состояние мужских половых клеток. Другими способами обнаружить хромосомные аномалии невозможно.
По результатам генетических исследований делаются выводы о возможности искусственного оплодотворения здоровой яйцеклетки при нормальной беременности и рождении ребенка без генетических отклонений. Только генетик после тщательного обследования может с большой долей вероятности сказать, разрешено ли зачатие посредством процедуры ИКСИ или ЭКО, когда весь процесс оплодотворения происходит in vitro.
Наиболее частыми патологиями являются синдром Клайнфельтера и Y-дисомия, и чем раньше будет определен кариотип плода, тем больше шансов на ЗГТ. Обнаружение мутации в гене CFTR указывает на муковисцидоз разной степени, который не только вызывает мужское бесплодие, но и делает процедуру ИКСИ непродуктивной. Однако и здесь возможны варианты, когда из общего количества сперматозоидов отбираются для оплодотворения исправные половые клетки, что гарантирует зачатие, нормально развивающуюся беременность и рождение ребенка.
Выводы
Самостоятельно назначить себе лечение нельзя, нельзя пользоваться советами друзей или народными рецептами — только специалист может назначить правильное лечение по результатам ваших анализов. Не рискуйте своим здоровьем и возможностью стать отцом — обратитесь за помощью к врачам. Если при нормально организованной половой жизни беременность не наступает несколько лет, партнерам следует пройти генетическую диагностику, которая выявит скрытые генетические мутации по мужской линии. Диагноз «генетическое мужское бесплодие» — это не приговор, а лишь поиск вариантов решения проблемы.
Генетически обусловленные формы бесплодия у мужчин: основные характеристики и практические аспекты лабораторной диагностики
Д.С. Михайленко1,2,3, И.Ю. Соболь1, Е.А. Ефремов1, О.И. Аполихин1, А.С. Танас3, Б.Я. Алексеев1, М.В. Немцов2.3
- 1 НИИ урологии и интервенционной радиологии им. VI СУ. Лопаткина — филиал Национального медицинского исследовательского центра радиологии Минздрава России; 105425, ул. 3 Парковая д. 51, Москва, Россия
- 2 ФГАУ ВО Первый Московский государственный медицинский университет имени М.В. ИМ. Сеченова, Минздрав России (Сеченовский университет); Россия, 119991, ул. Трубецкая, 8, корп. 2, Москва, Россия
- 3 ФГБУ «Медико-генетический научный центр»; 115478, ул. Москворечье, 1, Москва, Россия
Бесплодие у мужчин является актуальной проблемой современной андрологии и встречается в той или иной форме в 45% бесплодных браков . При диагностике мужского бесплодия (МБ) постепенно исключаются частые причины заболевания: травмы, варикоцеле, аномалии развития, системные заболевания, прием стероидов, инфекции мочеполовой системы, хронический стресс, иммунологические нарушения и другие патологические изменения. В остальных случаях бесплодие у человека может быть вызвано генетическими причинами в виде хромосомных аберраций (например, синдром Клайнфельтера) или точечных мутаций, включая как редкие моногенные заболевания (синдром Кальмана, синдромы Картагенера и другие), так и более частые случаи заболевания азооспермия, вызванная делециями локуса AZF или комбинацией мутаций CFTR [2,3]. В работе систематизированы данные о генетических нарушениях, приводящих к снижению фертильности у мужчин, и методы их определения. Основное внимание уделяется молекулярно-генетической диагностике известных форм бесплодия, связанных с частыми мутациями; Охарактеризована область применения высокопроизводительного секвенирования в исследовании МБ. Обзор предназначен для андрологов, генетиков, урологов и других специалистов, занимающихся лабораторной диагностикой мужского бесплодия.
МАТЕРИАЛЫ И МЕТОДЫ
При написании обзора мы использовали данные о генетических нарушениях со снижением фертильности у мужчин, опубликованные в базах данных PubMed, Электронной научной библиотеке Elibrary.ru и на сайтах профессиональных андрологических ассоциаций. Поиск в базах данных проводился по ключевым словам «ген», «мужское бесплодие», «мутация» и «полиморфизм». На первом этапе было найдено 102 источника не старше 5 лет, относящихся к теме обзора. Исключались тезисы конференций, короткие сообщения, дубликаты публикаций. Затем, исходя из актуальности данных, надежности источников, импакт-факторов журналов и последовательности изложения материала в рукописи, были непосредственно выбраны 49 статей в рецензируемых международных научных журналах и два практических руководства за цитату в обзоре.
ХРОМОСОМНЫЕ АБЕРРАЦИИ ПРИ СНИЖЕНИИ ПОВОДНОСТИ У МУЖЧИН
Большинство случаев МБ, связанных с анеуплоидией, представлены синдромом Клайнфельтера (СК).
По разным оценкам, частота сердечной недостаточности среди бесплодных мужчин в целом составляет около 3%, в когорте пациентов с необструктивной азооспермией — до 10%. Классическая форма СК обусловлена кариотипом (47, XXY) и составляет 85% случаев этого синдрома, характеризуется низким уровнем тестостерона, гипогонадизмом, гинекомастией; в других случаях — множественные анеуплоидии (48, XXYY; 48, XXXY; 49, XXXXY), которые выражаются в более тяжелых фенотипических проявлениях в раннем детстве с формированием наружных гениталий и умственной отсталостью или частичным увеличением копий фрагмента Х-хромосома (47, X, iXq, Y). Частота классической формы СК составляет 1: 600 новорожденных. В некоторых случаях наблюдается мозаичная форма СК с кариотипом (47, XXY / 46, XY) и стертой клинической картиной заболевания. В некоторых случаях у пациентов с СК могут быть дети с использованием вспомогательных репродуктивных технологий, в частности, сбора спермы с помощью TESE (микродиссекция семенников из яичек) и последующей интрацитоплазматической инъекции сперматозоидов — ICSI (интрацитоплазматическая инъекция сперматозоидов) [4]. При СН в зависимости от выраженности фенотипических проявлений выявляется олиго или азооспермия с гиалинизацией и фиброзом семенных протоков.
Большинство случаев СК возникает в раннем периоде полового созревания, и их диагностика проста, но у 10-20% пациентов наблюдается мозаичная форма СК со стертыми гипогонадальными симптомами [5]. В этих случаях в детском и подростковом возрасте возможна недостаточная диагностика приливов, и такие мужчины могут обратиться к андрологу по поводу бесплодия. Генетический диагноз СК состоит из кариотипирования; Для оптимизации протокола ведения таких пациентов также определяется концентрация тестостерона, лютеинизирующего и фолликулостимулирующего гормонов, пролактина, ингибина B и некоторых других.
Поскольку большое количество генов попадает в область хромосомной аберрации во время SC, трудно идентифицировать какие-либо гены-кандидаты, ответственные за фенотипические варианты проявления SC. Таким образом, анализ транскриптома пациентов с сердечной недостаточностью по сравнению со здоровыми людьми в контрольной группе показал более тысячи дифференциально экспрессируемых генов, десятки из которых имели биологические функции, прямо или косвенно связанные с формированием полового характера и сперматогенезом [6]. В редких случаях МБ может быть вызвано сбалансированными транслокациями — описано около 250 таких случаев, причем некоторые из них представлены множественными перестройками с 4 и 6 точками разрыва [7], робертсоновскими транслокациями, инверсиями. В последних случаях наиболее информативным является анализ методом мультилокусной FISH (флуоресцентной гибридизации in situ) [8]. Цитогенетически детерминированные формы МБ включают синдром де ла Шапеля, проявляющийся гипоспадией, микропенисом, неполным выпадением яичек. С формулой кариотипа (46, XX) пациенты имеют мужской фенотип и являются носителями гена Y, определяющего пол (SRY), который был перемещен с Y-хромосомы на X-хромосому или аутосомы. Генетический диагноз этого синдрома состоит из кариотипа и, в случае кариотипа (46, XX), последующей FISH или ПЦР маркеров STS для обнаружения SRY [9]. Принимая во внимание вышесказанное, кариотип является одним из основных рутинных тестов на бесплодие у мужчин, который позволяет диагностировать СК, исключая другие хромосомные аномалии. При необходимости его можно интегрировать с мультилокусным FISH для обнаружения небольших сбалансированных перестроек или ПЦР гена SRY.
УДАЛЕНИЕ ОБЛАСТИ AZF НА Y-ХРОМОСОМЕ
Субмикроскопические хромосомные аберрации (микроделеции) Y-хромосомы занимают лидирующее положение по частоте встречаемости как причина МБ, выявляемых молекулярно-генетическими методами. Микроделеции Y-хромосомы, влияющие на область AZF (фактора азооспермии), могут включать субрегионы AZFa, b и / c и приводить к потере генов USP9Y, DBY, UTY, TB4Y и других локусов, участвующих в регуляции сперматогенеза. Одновременные делеции субрегионов AZFa, b или нескольких субрегионов приводят к азооспермии; частичные делеции субрегиона AZFc могут приводить к олигозооспермии [10]. Особенностью Y-хромосомы является наличие 8 обратных повторов в ее длинном плече. Большинство делеций AZF вызвано несбалансированной неаллельной гомологичной рекомбинацией между последовательностями эндогенного ретровируса HERV15 (делеции AZFa), палиндромом P5 и проксимальным плечом палиндрома P1 (делеции AZFb); палиндромы P5 и P4, а также проксимальные и дистальные ветви палиндрома P1 (делеции AZFb + c), прямые повторы b2 и b4 (полные делеции AZFc).
Чаще всего встречаются делеции AZFc — 60-70% всех делеций AZF, на втором месте по частоте — делеции AZFb и b + c (15-20%), делеции AZFa встречаются реже (5-10%), в редких случаях — сложные обнаружены делеции AZFa + b, a + b + c [11, 12]. Частота делеции AZF у мужчин с бесплодием в России составляет около 10%, что близко к показателям в других странах Восточной Европы (8-12%); эта частота несколько ниже в азиатских популяциях (6–9%) [10–12]. Как правило, диагностика делеций AZF проводится с помощью ПЦР участков Y-хромосомы (по 2-3 маркера STS для каждой из подобластей AZF) в областях возможных делеций и контрольных маркеров одной и той же хромосомы. Особый интерес представляет определение частичных делеций AZFc, доля которых среди пациентов с олигозооспермией может достигать 12%; в этих случаях возможно получение потомства. При необходимости могут быть протестированы дополнительные маркеры STS Y-хромосомы, связанные с AZFc [13, 14]. Хотя минимальный набор маркеров STS, необходимых для диагностики делеций AZF, присутствует в каждой тест-системе и в целом соответствует международным рекомендациям, возможны различия в дополнительных маркерах STS, контрольных локусах Y-хромосомы, методах обнаружения (электрофорез продуктов мультиплексной ПЦР или обнаружение в режиме ПЦР в реальном времени). На рис. 1 представлены данные о составе некоторых систем обнаружения делеций AZF, используемых отечественными авторами как в прикладных исследовательских проектах, так и в диагностических целях 17. Следует отметить, что в ДНК-диагностике более целесообразным представляется использование тест-систем в режиме ПЦР в реальном времени, исходя из скорости получения результата и сложности этапов анализа.
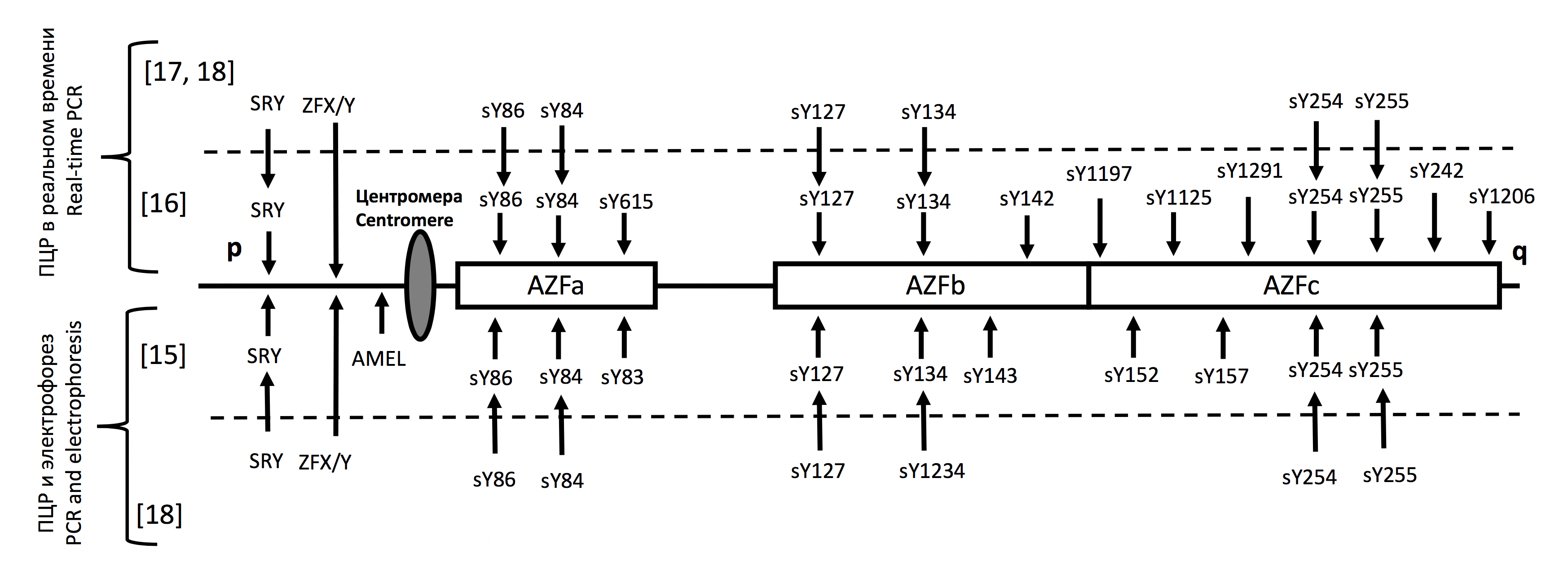
Рис. 1. Расположение маркеров STS в области AZF и контрольных локусов в различных тест-системах для определения микроделеций Y-хромосомы
ИНЖИР. 1. Локализация маркеров и контролей STS в области AZF, относящихся к различным тест-системам, предназначенным для обнаружения микроделеций Y
Мутации CFTR при бесплодии у мужчин
Ген CFTR кодирует трансмембранный регулятор проводимости, который участвует в транспорте ионов хлора через плазматическую мембрану и в образовании секреции эпителиальных клеток. Сложные гетерозиготы для тяжелых и легких мутаций CFTR не имеют клинической картины муковисцидоза, как у пациентов с инактивацией обоих аллелей. Однако у них есть проявления гаплонедостаточности — измененная концентрация ионов хлора и электролитов, повышенная вязкость секрета в малых протоках желез внешней секреции, что связано с агенезом семявыносящего протока (CBAVD — врожденное двустороннее отсутствие семявыносящего протока) семявыносящий проток) [19].
Пациентам с обструктивным МБТ показано генетическое тестирование мутаций CFTR. В этом случае тяжелую мутацию можно обнаружить с помощью тест-систем, предназначенных для поиска мутаций CFTR, часто встречающихся при диагностике муковисцидоза, которые включают до 18 (часто) диагностически значимых мутаций в европейской части России [20,21]. Вторая мягкая мутация в большинстве случаев — это аллель 5T. Это один из вариантов мононуклеотидного тракта IVS8-Tn, расположенного на границе интрон-экзон, который в 95% случаев представлен аллелями длиной 7 и 9T. Короткий аллель 5T приводит к нарушению сплайсинга, при котором вырабатывается только 30% нормального транскрипта CFTR. Сравнивая образцы от пациентов с CBAVD и контрольной группы кавказского происхождения, наблюдалось увеличение частоты встречаемости аллеля 5T: 3% против 21% и более [22, 23]. Обратите внимание, что не всегда возможно намеренно разделить пациентов с CBAVD на ранней стадии постановки диагноза. Однако, согласно недавним исследованиям, проведенным в российской популяции, частота аллеля 5T также повышена в гетерогенной выборке пациентов с МБ [15]. К такому же выводу приводит исследование 2146 российских мужчин с репродуктивными расстройствами, проведенное другой исследовательской группой [24]. Это делает целесообразным определение генотипа в локусе IVS8-Tn, также как часть генетического тестирования для первичной диагностики МБ.
Экспрессивность аллеля 5T зависит от повтора (TG) n, расположенного в непосредственной близости от IVS8-Tn. Было показано, что отсутствие синтеза нормальной изоформы белка CFTR при прочих равных обратно пропорционально длине повтора (TG) n. Гаплотип 5Т-13TG достоверно чаще ассоциируется с нарушением функции гена CFTR, чем 5Т-11TG в ряде популяций. В связи с этим целесообразно интегрировать генотипирование локуса IVS-8Tn путем определения длины повтора (TG) n [25]. Делеции AZF и сложных гетерозигот для мутаций CFTR являются наиболее частыми молекулярно-генетическими аномалиями, тестируемыми на MB. Положительный результат этих тестов позволяет установить причину МБ, в некоторых случаях скорректировать тактику ведения пациентов в клинике.
ПОЛИМОРФИЗМ AR И ДРУГИХ ГЕНОВ, СВЯЗАННЫХ С МУЖСКИМ БЕСПЛОДСТВОМ
Наряду с точечными мутациями некоторые авторы идентифицируют неблагоприятные аллели полиморфизма гена рецептора андрогена (AR) и генов семейства глутатионтрансфераз как возможные причины бесплодия у мужчин. Андрогены играют важную роль в сперматогенезе, реализуя свое действие через рецептор андрогенов. Первый экзон гена AR содержит полиморфный повтор CAG, соответствующий полиглутаминовому участку переменной длины. Длина повтора обратно пропорциональна способности рецептора активировать транскрипцию генов-мишеней, что может иметь важное значение в патогенезе бесплодия у носителей длинных аллелей [26]. Связь повторения AR CAG с бесплодием не была подтверждена во всех исследованиях, однако, согласно результатам недавнего метаанализа 40 оригинальных статей, в том числе 3858 мужчин с бесплодием и 3161 человек в контрольной группе, Было показано, что CAG повторяет аллели с мужским бесплодием в объединенной выборке: стандартизованная разница средних (SMD) составляла 0,14 с 95% доверительным интервалом 0,02–0,26. Популяционная ассоциация с длинными аллелями показана для популяций Европы и Северной Америки, но не для популяций Северной Африки или Юго-Восточной Азии. В метаанализе условно длинные аллели включали все повторы более чем 21 тринуклеотида CAG, то есть наиболее частый аллель [27]. Выбор пороговой длины повтора CAG напрямую влияет на результат этих исследований. С точки зрения повышения информативности анализа полиморфизма AR как предрасполагающего фактора МБ, некоторые авторы предлагают отнести более длинные повторы CAG26 к группе условно длинных аллелей [15]. Изучение полиморфизма АР у российских пациентов с МБ проводилось в разное время независимыми исследовательскими группами. Интересно, что одно из исследований показало ассоциацию сперматогенных нарушений с условно длинными (больше CAG28) и короткими (меньше CAG19) аллелями AR [27]. В целом общее количество исследованных выборок российских пациентов с МБ в имеющихся публикациях превышает 300 выборок; для них был получен результат, аналогичный наблюдаемому в других европейских популяциях: ассоциация условно длинных аллелей AR CAG-повтора с MB [15, 28, 29].
Гены GSTM1 и GSTT1 кодируют глутатион-трансферазы, ферменты, участвующие в детоксикации ксенобиотиков и в защите макромолекул от перекисного окисления. Гомозиготы по аллелям с делециями этих генов (нулевые генотипы) характеризуются повышенным риском развития ряда многофакторных заболеваний, в том числе МБ, по данным российских и зарубежных авторов [30, 31]. В частности, исследование на российских пациентах показало достоверное увеличение частоты нулевых генотипов в группе из 160 мужчин с бесплодием по сравнению со 104 выборками в контрольной популяции, в то время как частоты делеционных генотипов у пациентов и в контроле были взаимосвязаны до 50%, 37% против (GSTM1), 28% против 13% (GSTT1) и 19% против 5% (оба гена) [32]. Принимая во внимание вышесказанное, длинные аллели AR CAG-повтора с разумно выбранным уровнем отсечения и соединения для нулевых генотипов GSTM1 + GSTT1 можно рассматривать как генетические варианты зародышевой линии с низким проникновением, предрасполагающие к развитию МБ у мужчин европеоидной расы. Однако результаты генотипирования полиморфизмов сложно реализовать на практическом уровне: если обнаружение делеции AZF или гетерозиготного соединения CFTR позволяет установить причину бесплодия, то наличие неблагоприятных аллелей полиморфизмов говорит только о возможном генетическая предрасположенность к МБ.
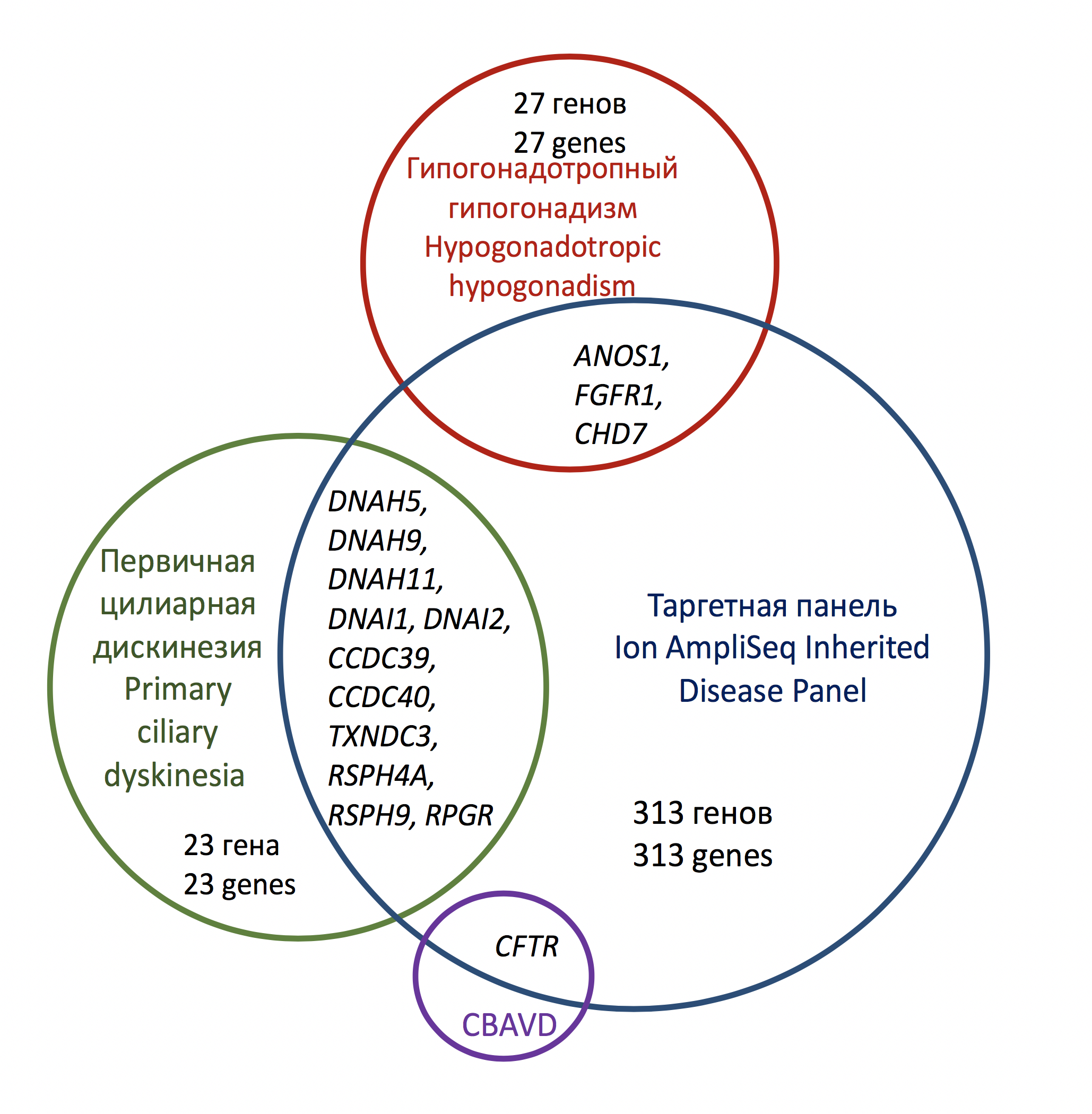
Рис. 2. Охват генов-кандидатов в основных моногенных формах МБ с помощью расширенной целевой панели AmpliSeq для NGS-диагностики наследственных заболеваний
ИНЖИР. 2. Диаграмма, показывающая гены, участвующие в развитии моногенных форм мужского бесплодия и охваченные комплексной панелью Ion AmpliSeq Inherited Disease Panel
МОНОГЕННЫЕ СИНДРОМАЛЬНЫЕ ФОРМЫ МУЖСКОГО БЕСПЛОДИЯ
В дополнение к типичному моногенному заболеванию, CBAVD с мутациями CFTR, были описаны другие синдромные формы MB, вызванные точечными мутациями в расширенных генах-кандидатах. Их особенность — генетическая гетерогенность, наличие нескольких десятков генов-кандидатов и разные типы наследования.
Врожденный гипогонадотропный гипогонадизм (ВГ) — это заболевание, при котором нарушается синтез, секреция или механизм действия в клетках-мишенях гонадотропин-рилизинг-гормона, основного гормона, регулирующего репродуктивную функцию человека. В результате не достигается полная половая зрелость и возникает бесплодие. Синдром характеризуется клинической и генетической неоднородностью (описано около 30 генов-кандидатов), помимо бесплодия, у 50% пациентов наблюдается аносмия (эта форма заболевания называется синдромом Калмана), у некоторых пациентов наблюдаются отклонения в развитии выявляются зубы, слуховые органы и скелет [33]. HH — это генетически гетерогенное заболевание, различные его формы наследуются по аутосомно-рецессивному, аутосомно-доминантному или X-сцепленному рецессивному типу. Первый ген-кандидат для заболевания был идентифицирован у пациента с синдромом Калмана — ANOS1, который кодирует аносмин, который участвует в сигнальном пути рецептора фактора роста фибробластов. Другие гены-кандидаты для синдрома Калмана, особенно при аутосомно-рецессивной форме заболевания, включают FGFR1, FGF8, CHD7, SOX10, SEMA3A, WDR11, HS6ST1, IL17RD, FEZF1, PROKR2 и PROK2, некоторые второстепенные гены-кандидаты [34]. Нормосмический HH может быть вызван мутациями в гене GNRHR (кодирующем гормон высвобождения гонадотропина), GNRH1, KISS1 и KISS1R, TAC3 и TACR3. При диагностике заболевания обращают внимание на крипторхизм и микропенис у мальчиков, которые проходят тестирование на гормоны (гонадотропины, половые стероиды, ингибин В). У подростков и в пожилом возрасте важно проводить дифференциальную диагностику с функциональными нарушениями и аденомой гипофиза, однако в этом возрасте клинические признаки становятся более выраженными, в том числе характерные для некоторых форм синдрома: аносмия у пациентов с мутациями ANOS1 , нарушения скелета у пациентов с мутациями FGFR1 или FGF8, колобома и болезни сердца и носители мутаций гена CHD7, нарушение слуха у пациентов с мутациями SOX10 [35,36].
Первичная цилиарная дискинезия (CD, синдром Картагенера, OMIM 244400) — врожденное заболевание с частотой 1:20 тысяч новорожденных, при котором функционирование клеточных ресничек значительно снижено или отсутствует. Клинические признаки заболевания, вызванного дисфункцией ресничек, включают хронические инфекции дыхательных путей с первого года жизни, бесплодие у мужчин и высокий риск внематочной беременности у женщин в более позднем возрасте; у некоторых пациентов наблюдаются врожденные пороки сердца, бронхоэктазы, пигментация сетчатки, поликистоз почек, патологические изменения структуры печени и пищевода. Заболевание характеризуется значительной клинической и генетической неоднородностью, понятной: существует 33 гена-кандидата на синдром Картагенера, мутации в каждом из них могут приводить к дисфункции ресниц и комбинированным фенотипическим изменениям. При лабораторной диагностике заболевания использовалась электронная микроскопия ресничек и мест их прикрепления, оценка уровня оксида азота в носу и т.д., но до недавнего времени специфической лабораторной диагностики первичной БК не было [37].
РОЛЬ ВЫСОКОЭФФЕКТИВНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ В АНАЛИЗЕ ГЕРМИНАЛЬНЫХ МУТАЦИЙ В СНИЖЕНИИ ФЕРТОВИМОСТИ У МУЖЧИН
В настоящее время можно выделить 78 генов, мутации в которых приводят к 92 различным фенотипическим вариантам MB [38]. За исключением небольшого числа случаев с крупными мутациями, диагностика идиопатического МБ требует секвенирования расширенных кодирующих областей генов-кандидатов, которое выполняется с использованием технологий высокопроизводительного секвенирования (NGS — секвенирование следующего поколения). Во-первых, NGS незаменим для секвенирования экзома, которое рекомендуется проводить у пациентов с МБ без диагностических признаков, характерных для ранее описанных форм заболевания. Например, секвенирование экзома выявило новую бессмысленную мутацию c.112C> T, p.R38 * в гене GNRHR у пациента с HH [39]. Другой пример: в первичной CD с использованием секвенирования экзома в гене LRRC6 была идентифицирована сложная гетерозигота: c.183T> G (p.N61K); c.179-1G> A) [40].
Однако секвенирование специально подобранной генетической панели кажется более подходящим с точки зрения взаимосвязи между стоимостью и клинической чувствительностью, чем анализ для диагностики моно- и олигогенных форм MB — CBAVD, GG и CD. Например, секвенирование 69 генов-кандидатов HH на платформе MiSeq (Illumina) выявило патогенную мутацию у 11 из 28 неродственных пациентов, то есть в 40% случаев, что является довольно высоким показателем для прямой ДНК-диагностики этого заболевания [41] . Секвенирование 32 первичных генов-кандидатов на CD у 46 пациентов с подозрением на заболевание позволило идентифицировать мутацию зародышевой линии у 10 из них с помощью технологии IonTorrent [42]. Достаточно высокий процент подтвержденных случаев врожденной БК наблюдался у 51 итальянского пациента. Секвенирование 24 генов-кандидатов с использованием технологии AmpliSeq на платформе IonTorrent выявило 5 гомозигот и 27 сложных гетерозигот, что вместе составляет 43% подтвержденных случаев [43]. Большая клиническая чувствительность была достигнута только при секвенировании уменьшенной версии экзома (клинического экзома) на платформе Illumina — мутации зародышевой линии были обнаружены у 52% пациентов (11/21) [44]; o при секвенировании обширных генетических панелей, включающих не десятки, а сотни генов: секвенирование кодирующих частей 284 генов, прямо или косвенно связанных с патогенезом CD, у 74 датских пациентов выявило патогенные генетические варианты зародышевой линии в 67% случаев [45]. Однако в составе таких больших панелей подавляющее большинство локусов не имеют отношения к патогенезу МБ и их амплификация избыточна как с генетической, так и с экономической точки зрения. Это можно проиллюстрировать охватом генов-кандидатов для моногенных форм МБ в модифицированной теперь панели Ion AmpliSeq ™ Inherited Disease Panel (IonTorrent), которая включает 328 генов (рис. 2). Панель содержала 95% избыточных генов и только 3 из 30 генов-кандидатов на гипогонадотропный гипогонадизм. Доля избыточных генов в панели TruSight Inherited Disease Sequencing Panel (Illumina, содержит 552 гена), которая теперь предлагается конечным пользователям от другого производителя, еще больше [46,47]. В недавнем обзоре 23 оригинальных исследований нарушений фертильности у мужчин с использованием технологий NGS, а также в некоторых других работах, некоторые авторы соглашаются с тем, что для диагностики МБ было бы актуально использовать специальную целевую панель, объединяющую различные гены-кандидаты снижение фертильности у мужчин [48.49]. Такая панель генов NGS для диагностики MB может включать гены врожденных форм GH и CD, ген CFTR и другие гены-кандидаты MB, идентифицированные во время секвенирования экзома мужчин с нарушенной фертильностью.
ЗАКЛЮЧЕНИЕ
Таким образом, генетическая патология ответственна за значительную часть МБ и представлена различными генетическими нарушениями: хромосомными аберрациями в виде анеуплоидий и делеций разной длины, точечными мутациями в кодирующих частях генов и генетическими вариантами с низким уровнем проникновения. В большинстве случаев МБ с предположительно генетическими причинами проводится кариотип для исключения хромосомных аберраций, проводится анализ делеций AZF, в случае обструктивных МБ проводится анализ частых мутаций CFTR. Генотипирование полиморфизмов (повтор AR CAG, гены семейства GST) в настоящее время не имеет диагностической ценности. И наоборот, технологии NGS приобретают все большее значение: оптимальный метод прямой ДНК-диагностики моно- и олигогенных форм MB в рамках одного анализа — это секвенирование кодирующих частей соответствующих генов-кандидатов как части специально подобранной панели-мишени. Обоснованное направление пациента на тот или иной вид генетического анализа, основанное на минимальных диагностических критериях известных синдромных форм МБ, существенно повышает информативность лабораторной диагностики бесплодия у мужчин.
ЛИТЕРАТУРА
- Чалый М.Е., Ахвледиани Н.Д., Харчилава Р.Р. Мужское бесплодие. Урология 2017 (2, приложение 2): 4-19 doi: 10.18565 / urol. 2017.2-приложение. 4-19. [Чалый М.Е., Ахвледиани Н.Д., Харчилава Р.Р. Мужское бесплодие. Урология = Урология 2017; (2, доп.): 4-19. (На русском)]
- Кляйш С. Диагностика мужского бесплодия: диагностическое обследование бесплодного мужчины. Eur Urol Suppl 2014; 13: 73-82 DOI: 10.1016 / j.eursup.2014.08.002.
- Краус Ч., Риера-Эскамилла А. Генетика мужского бесплодия. Nat Rev Urol 2018; 15 (6): 369-84 DOI: 10.1038 / s41585-018-0003-3.
- Сираиси К., Мацуяма Х. Синдром Клайнфельтера: от педиатрии к гериатрии. Reprod Med Biol 2018; 18 (2): 140-50 DOI: 10.1002 / rmb2.12261.
- Бономи М., Рочира В., Паскуали Д., Балерсия Дж., Джаннини Е.А., Ферлин А и др. синдром Клайнфельтера (СК): генетика, клинический фенотип и гипогонадизм. J Endocrinol Invest 2017; 40 (2): 123-34 DOI: 10.1007 / s40618-016-0541-6. PMID: 27644703.
- Хоксворт Д. Д., Шафран А. А., Джордан П. У., Добс А. С., Герати А. С.. Бесплодие у пациентов с синдромом Клайнфельтера: оптимальные сроки криоконсервации спермы и ткани яичек. Rev Urol 2018; 20 (2): 56-62 DOI: 10.3909 / riu0790.
- Nguyen MH, Morel F, Pennamen P, Parent P, Douet-Guilbert N, Le Bris MJ и др. сбалансированная комплексная хромосомная перестройка при мужском бесплодии: клинический случай и обзор литературы. Андрология 2015; 47 (2): 178-85 DOI: 10.1111 / e.12245.
- Chatziparasidou A, Christoforidis N, Samolada G, Nijs M. Анеуплоидия сперматозоидов у бесплодных пациентов мужского пола: систематический обзор литературы. Андрология 2015; 47 (8): 847-60 DOI: 10.1111 / e.12362.
- Маджзуб А., Арафа М., Старкс С., Эльбардиси Х., Аль-Саид С., Сабанег Э. 46 ХХ кариотип при оценке мужской фертильности; серия случаев и обзор азиатской литературы J Androl 2017; 19 (2): 168-72 DOI: 10.4103 / 1008-682X.181224.
- Колако С., Моди Д. Генетика Y-хромосомы человека и ее связь с мужским бесплодием. Репрод Биол Эндокринол 2018; 16 (1): 14 DOI: 10.1186 / s12958-018-0330-5.
- Лю XG, Ху HY, Го YH, Sun YP. Корреляция между микроделецией Y-хромосомы и мужским бесплодием. Genet Mol Res 2016; 15 (2): gr 15028426 doi: 10.4238 / gmr.15028426.
- В.Б. Черных делеции AZF — частая генетическая причина мужского бесплодия: современное состояние дел. Выпуски репродукции 2009 г .; (1): 10-15. [Черных В.Б. Делеции AZF — частая генетическая причина мужского бесплодия: современное состояние исследований. Проблемы репродукции = Проблемы с воспроизведением 2009 г .; (1): 10-5. (на русском)]
- Nailwal M, Chauhan JB. Субрегион фактора азооспермии Y хромосомы. J Hum Reprod Sci 2017; 10 (4): 256-60 doi: 10.4103 / jhrs. JHRS_16_17.
- Сафина Н.Ю., Яманди Т.А., Черных В.Б., Акуленко Л.В., Боголюбов С.В., Витязева И.И и другие генетические факторы мужского бесплодия, их сочетания и характеристики спермы мужчин с нарушенной фертильностью. Андрология и генитальная хирургия 2018; 19 (2): 40-51 .doi: 10.17650 / 2070-9781-2018-19-2-40-51. [Сафина Н.Ю., Яманди Т.А., Черных В.Б., Акуленко Л.В., Боголюбов С.В., Витязева И.И и др. сенетические факторы мужского бесплодия, их сочетания и сперматологические характеристики мужчин с дефицитом фертильности. Андрология и генитальная хирургия = Андрология и генитальная хирургия 2018; 19 (2): 40-51. (На русском)]
- Михайленко Д.С., Соболь И.Ю., Сафронова Н.Ю., Симонова О.А., Ефремов Е.А., Ефремов Г.Д и др. састота выявления делеций AZF, мутаций CFTR и длинных аллелей повтора AR CAG при первичной лабораторной диагностике в гетерогенная группа пациентов с мужским бесплодием. Урология 2019; (3): 101-7 doi: 10.18565 / urology.2019.3.101-107 [Михайленко Д.С., Соболь И.Ю., Сафронова Н.Ю., Симонова О.А., Ефремов Е.А., Ефремов Г.Д и др. састота делеций AZF, мутаций CFTR и длинных аллелей AR CAG-повторов при первичной лабораторной диагностике в гетерогенной группе бесплодных мужчин. Урология = Урология 2019; 3: 101-7. (На русском)]
- Делеции локуса AZF. URL: https://www.dna-technology.ru/sites/default/ files / azf_051-3.pdf [Отмена AZF. URL: https://www.dna-technology.ru/ sites / default / files / azf_051-3.pdf (на русском языке)]
- Аксельрод Е.В., Миронов К.О., Михайленко Д.С., Ефремов Г.Д., Перепечин Д.В., Алексеев Б.Я и др. сазработка и апробация методики определения клинически значимых микроделеций в хромосоме Y на основе мультиплексной полимеразной цепной реакции в режиме реального времени. Лаборатория клинической диагностики. 2018; 63 (2): 124-8 DOI: 10.18821 / 0869-2084-2018-63-2-124-128. [Аксельрод Э.В., Миронов К.О., Михайленко Д.С., Ефремов Г.Д., Перепечин Д.В., Алексеев Б.Ю и др. сазработка и утверждение методики на основе мультиплексной полимеразной цепной реакции в реальном времени для определения клинически значимых микроделеций в хромосоме Y. Клиническая лабораторная диагностика = Клиническая лабораторная диагностика 2018; 63 (2): 124-8. (На русском)]
- Набор реагентов для обнаружения микроделеций в локусе AZF хромосомы Y в геноме человека методом ПЦР. URL: http://www.lytech.ru/ product / genetika-cheloveka / mikrodeletsii-azf-lokusa-y-khromosomy / azf-deletions /. [Набор для обнаружения микроделеций Y-хромосомы в локусах AZF человека с помощью ПЦР. URL: http://www.lytech.ru/ product / genetika-cheloveka / mikrodeletsii-azf-lokusa-y-khromosomy / azfdeletions /. (На русском)]
- Лю XG, Ху HY, Го YH, Sun YP. Корреляция между микроделецией Y-хромосомы и мужским бесплодием. Genet Mol Res 2016; 15 (2): gr 15028426 doi: 10.4238 / gmr.15028426.
- Никифорова А.И., Абрамов Д.Д., Зобкова Г.Ю., Горяинова А.В., Семыкин С.Ю., Шубина Э и др. спределение мутаций гена CFTR у детей с муковисцидозом. Вестник РГМУ 2018; 3: 35-41 DOI: 10.24075 / vrgmu.2018.037. [Никифорова А.И., Абрамов Д.Д., Зобкова Г.Ю., Горяинова А.В., Семыкин С.Ю., Шубина Ю и др. сыявление мутаций CFTR у детей с муковисцидозом. Вестник РГМУ = Вестник РГМУ. 2018; 3: 35-41. (На русском)]
- Черных В.Б., Степанова А.А., Бескоровая Т.С., Сорокина Т.М., Шилейко Л.В., Курило Л.Ф и др. састота и спектр мутаций и полиморфизм IVS8-T гена CFTR у российских мужчин с генетическим бесплодием 2010; 46 (6): 844-52. [Черных В.Б., Степанова А.А., Бескоровая Т.С., Сорокина Т.М., Шилейко Л.В., Курило Л.Ф и др. састота и спектр мутаций и полиморфизм IVS8-T гена CFTR у бесплодных мужчин России. Genetics = Genetics 2010; 46 (6): 750-7. (На русском)]
- Ян Х, Сун Цюй, Юань П, Лян Х, Ву Х, Лай Л и др. совые мутации и полиморфизмы в гене CFTR связаны с тремя подтипами врожденного отсутствия семявыносящего протока. Fertil Steril 2015; 104 (5): 1268-75.e1-2 DOI: 10.1016 / j.fertnstert.2015.07.1143.
- Гельфи С., Перего М., Ригетти П.Г., Кайнарка С., Фирпо С., Феррари М и др. сыстрый электрофорез капиллярной зоны в изоэлектрическом гистидиновом буфере: высокое разрешение аллельных вариантов поли-Т тракта в 8-м интроне гена CFTR. Clin Chem 1998; 44 (5): 906-13.
- Соловьева Е.В., Татару Д.А., Преда О.Г., Артюхова В.Г., Секира А.Г., Деревьева В.Ю и др. сутации гена CFTR у мужчин с бесплодием. Медицинская генетика 2018; 17 (5): 28-38 DOI: 10.25557 / 2073-7998.2018.05.28-38. [Соловьева Е.В., Татару Д.А., Преда О.Г., Артюхова В.Г., Секира А.Г., Деревьева В.Ю и др. сутации CFTR при мужском бесплодии. Медицинская генетика = Медицинская генетика 2018; 17 (5): 28-38. (На русском)]
- Салинас ДБ, Азен С., Янг С., Кинз Т.Г., Харрази М, Парад РБ. Фенотипы младенцев с МВ в Калифорнии положительны при скрининге аллеля CFTR 5T на длину повтора TG. Genet Test Mol Biomarkers 2016; 20 (9): 496-503 DOI: 10.1089 / gtmb.2016.0102.
- Готтлиб Б., Ломброзо Р., Бейтель Л.К., Трифиро М.А. Молекулярная патология рецептора андрогенов при мужском бесплодии. Reprod Biomed Online 2005; 10 (1): 42-8.
- Сяо Ф, Лан А, Линь З, Сонг Дж, Чжан И, Ли Дж и др. слияние длины CAG-повтора в гене рецептора андрогенов на мужское бесплодие — метаанализ. Играть в BioMed Online 2016; 33 (1), 39-49 DOI: 10.1016 / j.rbmo.2016.03.012.
- Черных В.Б., Руднева С.А., Сорокина Т.М., Шилейко Л.В., Остроумова Т.В., Ермолаева С.А и др. слияние CAG-полиморфизма гена рецептора андрогенов (АР) на сперматогенез у мужчин с бесплодием. Андрология и генитальная хирургия 2015; 16 (4): 55-61 DOI: 10.17650 / 2070-9781-2015-16-4-55-61. [Черных В.Б., Руднева С.А., Сорокина Т.М., Шилейко Л.В., Остроумова Т.В., Ермолаева С.А и др. слияние гена СAG-полиморфизма рецептора андрогенов (AR) на сперматогенез у бесплодных мужчин. Андрология и генитальная хирургия = Андрология и генитальная хирургия 2015; 16 (4): 55-61. (На русском)]
- Михайленко Д.С., Бабенко О.В., Кириллова Е.А., Никифорова О.К., Зарецкая Н.В., Курносова Т.Р и др. сомплексный молекулярно-генетический анализ микроделеций AZF, мутаций гена CFTR и длины CAG-повтора гена AR у мужчин с бесплодием. Выпуски репродукции 2005 г .; 11 (6): 52-5. [Михайленко Д.С., Бабенко О.В., Кириллова Е.А., Никифорова О.К., Зарецкая Н.В., Курносова Т.Р и др. сомплексный молекулярно-генетический анализ микроделеций AZF, мутаций CFTR и длины CAG-повторов гена AR у бесплодных мужчин. Проблемы репродукции = Проблемы воспроизводства 2005; 11 (6): 52-5. (На русском)]
- Глыбочко П.В., Аляев Ю.Г., Чалый М.Е., Усачева О.А. Влияние полиморфизма генов глутатионтрансферазы М1 и Т1 на андрогенное состояние и качество эякулята после хирургического лечения варикоцеле. Андрология и генитальная хирургия 2013, 14 (1): 23-26. [Глыбочко П.В., Аляев Ю.Г., Чалый М.Е., Усачева О.А. Влияние полиморфизмов генов глутатион-S-трансферазы Т1 и М1 на андрогенный статус и качество спермы после хирургического лечения варикоцеле. Андрология и генитальная хирургия = Андрология и генитальная хирургия 2013; 14 (1): 23-26. (на русском)]
- Wu W, Lu J, Tang Q, Zhang S, Yuan B, Li J и др. сулевые полиморфизмы GSTM1 и GSTT1 и риск мужского бесплодия: обновленный метаанализ, включающий 6934 субъекта. Ski Rep 2013; 3: 2258 DOI: 10.1038 / srep02258.
- Курашова Н.А., Беляева Е.В., Ершова О.А., Дашиев Б.Г., Баирова Т.А., Колесникова Л.И. Связь полиморфизма генов GSTT1, GSTM1 с бесплодием у мужчин. Урология 2017; (6): 38-42 DOI: 10.18565 / urology. 2017.6.38-42. [Курашова] Н.А., Беляева Е.В., Ершова О.А., Дашиев Б.Г., Баирова Т.А., Колесникова Л.И. Связь полиморфизма генов GSTT1 и GSTM1 с бесплодием у мужчин. Урология = Урология 2017; 6: 38-42. (На русском)
- Бем У., Булу П.М., Даттани М.Т., де Ру Н., Доде С., Дункель Л и др. свропейское консенсусное заявление о врожденном гипогонадотропном гипогонадизме — патогенез, диагностика и лечение. Нат Рев эндокринол 2015; 11 (9): 547-64 DOI: 10.1038 / nrendo.2015.112.
- Стаму М.И., Георгопулос Н.А. Синдром Каллмана: фенотип и генотип гипогонадотропного гипогонадизма. Метаболизм 2018; 86: 124-34 DOI: 10.1016 / j.metabol.2017.10.012.
- Топалоглу АК. Обновленная информация о генетике идиопатического гипоганадотропного гипогонадизма. J Clin Res Pediatr Endocrinol 2017; 9 (Приложение 2): 113-22 DOI: 10.4274 / jcrpe.2017.S010. PMID: 29280744.
- Топалоглу А.К., Котан ЛД. Генетика гипогонадотропного гипогонадизма. Endocr Dev 2016; 29: 36-49 DOI: 10.1159 / 000438841.
- Мирра В., Вернер С., Сантамария Ф. Первичная цилиарная дискинезия: обновленная информация о клинических аспектах, генетике, диагностике и будущих стратегиях лечения. Front Pediatr 2017; 5: 135 DOI: 10.3389 / fped.2017.00135.
- Уд М.С., Волозонока Л., Смитс Р.М., Виссерс Л.Е., Рамос Л., Велтман Дж. Систематический обзор и оценка стандартизированной клинической валидности генов мужского бесплодия. Репродукция Человека 2019, 34 (5): 932-41 DOI: 10.1093 / humrep / dez022.
- Hussain HM, Murtaza G, Jiang X, Khan R, Khan M, Kakakhel MB, et al. Полное секвенирование экзома выявило новый бессмысленный вариант гена GNRHR, который вызывает нормосмический гипогонадотропный гипогонадизм в пакистанской семье. Horm Res Pediatr 2019; 91 (1): 9-16 DOI: 10.1159 / 000497114.
- Лю Л., Луо Х. Секвенирование всего экзома выявило новую сложную гетерозиготную мутацию LRRC6 у пациента с китайской первичной цилиарной дискинезией. Biomed Res Int 2018; 2018: 1854269 DOI: 10.1155 / 2018/1854269.
- Ким Дж. Х., Со Дж. Х., Ким Дж. Х., Ха Дж., Хван ИТ, Джанг Дж. Х и др. секвенирование целевой панели генов для молекулярной диагностики синдрома Каллмана и нормосмического идиопатического гипогонадотропного гипогонадизма. Exp Clin Endocrinol Diabetes 2019; 127 (8): 538-44 DOI: 10.1055 / a-0681-6608.
- Такеучи К., Китано М., Киётоши Х., Икегами К., Огава С., Икэдзири М и др. санель целевого секвенирования нового поколения выявляет новые мутации у японских пациентов с первичной цилиарной дискинезией. Аурис Насус Гортань 2018; 45 (3): 585-91 DOI: 10.1016 / j.anl.2017.09.007.
- Боаретто Ф., Снайдерс Д., Сальворо С., Спаллетта А., Мостаччуоло М.Л., Коллура М и др. сиагностика первичной цилиарной дискинезии с использованием целевой панели секвенирования нового поколения: молекулярные и клинические результаты у итальянских пациентов. J Mol Diagn 2016; 18 (6): 912-22 DOI: 10.1016 / j.jmoldx.2016.07.002.
- Анджелкович М., Минич П., Врека М., Стожилкович М., Скакич А., Совтик А и др. сеномный профиль поддерживает диагноз первичной цилиарной дискинезии и выявляет новые гены-кандидаты и генетические варианты. PLoS One 2018; 13 (10): e0205422 doi: 10.1371 / journal.pone.0205422.
- Пафф Т., Куи И.Е., Мутауакил Ю., Ризебос Э., Систерманс Е.А., Дэниэлс Х.Дж и др. сиагностическая ценность целевой панели генов у пациентов с первичной цилиарной дискинезией. Hum Mutat 2018 39 (5): 653-65 DOI: 10.1002 / humu.23403.
- Список целевых генов панели Ion AmpliSeq ™ по наследственным заболеваниям. URL: https://tools.thermofisher.com/content/sfs/brochures/CO25570_Ion_Inherited_Disease_GeneList_Table_CO25570.pdf
- Панель TruSight для определения последовательности наследственных заболеваний. URL: https: // emea.illumina.com/content/dam/illumina-marketing/documents/products/gene_lists/gene_list_trusight_inherited_disease.xlsx
- Робай А., Аббаси С., Акил А., Эль-Бардиси Х., Арафа М., Кристал Р.Г и др. систематический обзор генетики мужского бесплодия в эпоху секвенирования следующего поколения. Араб Дж Урол 2018; 16: 53-64 DOI: 10.1016 / j.aju.2017.12.003.
- Лаан М. Систематический обзор моногенетических причин мужского бесплодия: первый шаг к диагностическим генетическим панелям в андрологической клинике. Hum Reprod 2019, 34 (5): 783-5 DOI: 10.1093 / humrep / dez024.
Статья опубликована в журнале «Экспериментальная и клиническая урология» n. 1 2020, стр. 96-104